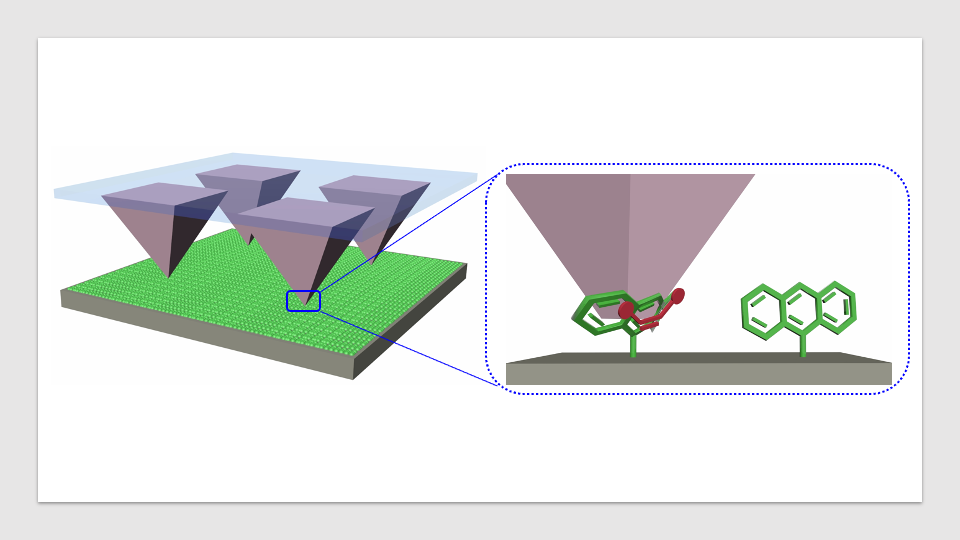
یک جریان کاری خودکار جدید توسعه یافته توسط دانشمندان در آزمایشگاه ملی لارنس برکلی (Berkeley Lab) قابلیت تجزیه و تحلیل محصولات آزمایشات واکنشهای آنها را به صورت زمان واقعی فراهم میکند، یک قابلیت کلیدی که برای فرآیندهای شیمیایی خودکار آینده ضروری است.
جریان کاری توسعه یافته – که تحلیل آماری را برای پردازش دادهها از طیفسنجی رزونانس مغناطیسی هستهای (NMR) اعمال میکند – میتواند به افزایش سرعت کشف داروهای جدید کمک کند و فرآیند توسعه واکنشهای شیمیایی جدید را شتاب بخشد.
دانشمندان آزمایشگاه برکلی که تکنیک نوآورانه را توسعه دادهاند میگویند که این جریان کاری به سرعت میتواند ساختار مولکولی محصولات تولید شده توسط واکنشهای شیمیایی را که پیشتر مورد مطالعه قرار نگرفتهاند، شناسایی کند. آنها اخیراً یافتههای خود را در مجله اطلاعات و مدلسازی شیمیایی (Journal of Chemical Information and Modeling) گزارش دادهاند.
علاوه بر کشف داروها و توسعه واکنشهای شیمیایی، این جریان کاری میتواند به دانشمندان کمک کند که در حال توسعه کاتالیزورهای جدید هستند. کاتالیزورها موادی هستند که فرآیند شیمیایی را در تولید محصولات جدید مفید مانند سوختهای تجدیدپذیر یا پلاستیکهای قابل تجزیه تسهیل میکنند.
“چیزی که بیشترین هیجان را در مردم ایجاد میکند، قابلیت آنچه این تکنیک میتواند از تجزیه و تحلیل واکنشهای زمان واقعی فراهم کند، است که بخشی جزئی از شیمی خودکار است”، گفته است که اولین نویسنده Maxwell C. Venetos، یک محقق سابق در بخش علوم مواد آزمایشگاه برکلی و دانشجوی دانشکده علوم مواد در دانشگاه کالیفرنیای برکلی بوده است. او در سال گذشته تحصیلات دکتری خود را به پایان رسانده است. “جریان کاری ما به شما اجازه میدهد واقعیت ناشناخته را دنبال کنید. شما دیگر به چیزهایی که جواب آنها را قبلاً میدانید محدود نیستید.”
جریان کاری جدید همچنین قادر است ایزومرها را شناسایی کند، که مولکولهایی با همان فرمول شیمیایی اما ترتیبات اتمی متفاوت هستند. این میتواند فرآیندهای شیمیایی سنتزی در تحقیقات داروسازی را به شدت شتاب دهد، به عنوان مثال. “این جریان کاری اولین نوع خود است که کاربران میتوانند کتابخانه خود را ایجاد کنند و آن را به کیفیت آن کتابخانه تنظیم کنند، بدون اتکا به پایگاه داده خارجی”، Venetos گفت.
پیشرفت برنامههای کاربردی جدید
در صنعت داروسازی، توسعه دهندگان داروها در حال حاضر از الگوریتمهای یادگیری ماشین برای پردازش مجازی صدها ترکیب شیمیایی استفاده میکنند تا نامزدهای جدید پتانسیلی را که احتمالاً در برابر س
رطانهای خاص و بیماریهای دیگر موثرتر هستند، شناسایی کنند. این روشهای انتخابی از کتابخانههای آنلاین یا پایگاه دادههای معروف ترکیبهای شناخته شده (یا محصولات واکنش) را جستجو میکنند و آنها را با “هدفهای” دارویی احتمالی در دیوارههای سلولی مطابقت میدهند.
اما اگر پژوهشگر دارو مولکولهایی را تجربه کند که ساختارهای شیمیایی آنها هنوز در یک پایگاه داده وجود ندارد، معمولاً باید روزها را در آزمایشگاه صرف تفکیک ترکیبات مولکولی از طریق دستگاه تصفیه، و سپس استفاده از یکی از ابزارهای مفید ترین شیمیدان مصنوعی، یک طیفسنج هستهای مغناطیسی (NMR)، برای شناسایی و اندازهگیری مولکولها در مخلوط یکییک صرف کند.
“اما با جریان کاری جدید ما، میتوانید به آسانی کلیه این کارها را در چندین ساعت انجام دهید”، Venetos گفت. صرفهجویی در زمان از توانایی جریان کاری برای تحلیل طیفهای NMR مخلوطهای واکنشی بدون تصفیه که حاوی ترکیبات چندگانه هستند، به وجود میآید، کاری که از طریق روشهای معمول تحلیل طیفهای NMR غیر ممکن است.
“من بسیار هیجانزده از این کار هستم چون این روشها را به روشهای نوآورانه دادهای در حل مشکل قدیمی شتاب میدهد”، گفته است که نویسنده سنیور Kristin Persson، یک دانشمند ارشد همکار در بخش علوم مواد آزمایشگاه برکلی و استاد دانشگاه کالیفرنیای برکلی در زمینه علوم و مهندسی مواد است که همچنین پروژه مواد را رهبری میکند.
نتایج تجربی
علاوه بر این که از روشهای تصفیه میزبان سریعتر هستند، جریان کاری جدید دارای پتانسیلی مشابه دقت است. آزمایشات شبیهسازی NMR انجام شده با استفاده از مرکز محاسبات علمی تحقیقاتی انرژی ملی (NERSC) در آزمایشگاه برکلی با حمایت از پروژه مواد نشان داد که جریان کاری جدید میتواند به درستی مولکولهای ترکیبات در مخلوطهای واکنشی که ایزومرها تولید میکنند را شناسایی کند و همچنین غلظتهای نسبی این ترکیبات را پیشبینی کند.
برای اطمینان از دقت آماری بالا، تیم تحقیق از الگوریتم پیشرفتهای به نام زنجیره مارکو وزنی هامیلتونی (HMCMC) برای تحلیل طیفهای NMR استفاده کردند. همچنین محاسبات نظری پیشرفتهای بر اساس روشی به نام نظریه چگالی-کارکردی (DFT) انجام دادند.
Venetos جریان کاری خود را به صورت متن باز طراحی کرده است تا کاربران بتوانند آن را بر روی یک رایانه میزکار عادی اجرا کنند. این راحتی برای هر کسی از صنعت یا دانشگاه مفید خواهد بود.
این تکنیک از مکالمات بین گروه Persson و همکاران آزمایشی Masha Elkin و Connor Delaney، پژوهشگران پست دکتری سابق در
گروه John Hartwig در دانشگاه کالیفرنیا در برکلی نشأت گرفته است. الکین اکنون استاد شیمی در موسسه فناوری ماساچوست، و دلانی استاد شیمی در دانشگاه تگزاس در دالاس است.
“در توسعه واکنش شیمیایی، ما به طور مداوم زمان را صرف میکنیم تا بفهمیم واکنش چه چیزی را ساخته و به چه نسبتی”، گفته است که John Hartwig، یک دانشمند ارشد همکار در بخش علوم شیمیایی آزمایشگاه برکلی و استاد شیمی در دانشگاه کالیفرنیا در برکلی است. “روشهای خاص طیفسنجی NMR دقیق هستند، اما اگر کسی در حال حاضر با مخلوطهای واکنشی خامی راه بیفتد که شامل محصولات بالقوه ناشناختهای باشد، این روشها برای یک جریان کاری آزمایشی یا خودکار با سرعت بالا بسیار زیاد خواهد شد. و اینجاست که این قابلیت جدید برای پیشبینی طیف NMR میتواند کمک کند”، او گفت.
اکنون که قابلیت جریان کاری خودکار را نشان دادهاند، Persson و تیم امیدوارند آن را در یک آزمایشگاه خودکار که طیفهای NMR هزاران یا حتی میلیونها واکنش شیمیایی جدید را در یک زمان تجزیه و تحلیل میکند، گنجانده شود.
.Materials provided by DOE/Lawrence Berkeley National Laboratory. Original written by Theresa Duque. Note: Content may be edited for style and length