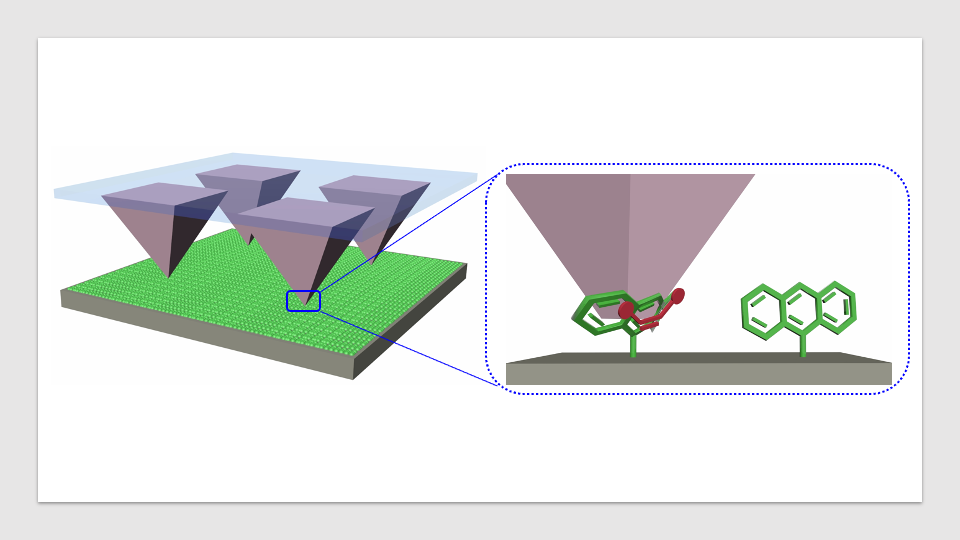
شیمیدانان اغلب واکنش های شیمیایی جدید را با استفاده از سیستم هایی به نام مدل، یعنی بسترهای ساده و به راحتی در دسترس، توسعه داده و بهینه می کنند. سپس از حدود 100 بستر دیگر به عنوان نمونه استفاده می کنند تا نشان دهند که واکنش کار می کند. این نمایش کاربرد همه کاره در اصطلاحات فنی «گستره» نامیده می شود. با این حال، انتخاب ذهنی از بسترها اغلب منجر به تصویری تحریف شده از دامنه کاربردهای واکنش تازه توسعه یافته می شود. اغلب مشخص نیست که آیا می توان از آن برای سنتز یک محصول مورد نظر استفاده کرد یا خیر. برای حل این مشکل، تیمی به رهبری شیمیدان پروفسور فرانک گلوریوس از دانشگاه مونستر (آلمان) روشی به کمک کامپیوتر و بدون تعصب برای انتخاب بسترهای مدل برای ارزیابی واکنشهای شیمیایی جدید پیشنهاد میکنند.
انتخاب بسترها بر اساس پیچیدگی و خواص ساختاری ترکیبات دارویی واقعی است. فرانک گلوریوس توضیح می دهد: “روش ما با هدف بهبود کیفیت و محتوای اطلاعاتی داده های واکنش شیمیایی در آینده و رفع شکاف های دانشی است.” درک عمیقتر واکنشهای جدید، موانع کاربرد آنها را در هر دو زمینه دانشگاهی و صنعتی کاهش میدهد. در دسترس بودن داده های باکیفیت و بی طرفانه نیز به طور قابل توجهی استفاده از یادگیری ماشین را تسهیل می کند و راه را برای استفاده جامع تر از داده ها هموار می کند.
به گفته نویسندگان این تیم، تلاش برای استانداردسازی و عینی سازی توسعه و ارزیابی واکنش های شیمیایی هنوز کاملا جدید و نسبتاً غیر معمول است. مایلیم با انتشار خود یک «فرایند بازاندیشی» را آغاز کنیم. نویسنده اول دبانجان رعنا می گوید.
دانشمندان دیگر نیز سعی کرده اند واکنش های شیمیایی را بر اساس بسترهای انتخاب شده “بهتر” ارزیابی کنند. با این حال، این کار به موارد خاص محدود شد – یا به ساختارهای کاملاً انتخاب شده با ارتباط دارویی یا به ساختارهایی که به طور خاص برای یک واکنش منفرد طراحی شده بودند، که باید در یک فرآیند پیچیده محاسبه و انتخاب شوند. برخلاف کار قبلی، روش ارائه شده توسط تیم مونستر، کل ساختار یک مولکول را در نظر می گیرد، که آن را به طور جهانی برای هر واکنش شیمیایی قابل استفاده می کند.
نیکلاس هولتر، یکی از نویسندگان مقاله در مونستر، فرآیند فکری پشت این مطالعه را توضیح میدهد: “محدوده در همه انتشارات مربوط به سنتز شیمیایی از اهمیت اساسی برخوردار است. با این حال، شیمیدانان اغلب در انتخاب ترکیبات زیرلایه برای آزمایش مغرضانه هستند. آنها بسترهایی را انتخاب می کنند که از نظر ساختاری بسیار ساده، بسیار شبیه به بستر مدل هستند یا به سادگی در آزمایشگاه در دسترس هستند (“تعصب انتخابی” آنها اغلب در انتشارات خود به هیچ وجه به واکنش های ناموفق اشاره نمی کنند تا تصویر بهتری ارائه دهند. “سوگیری گزارشی”).
هنگام سنتز ترکیبات شیمیایی جدید، مانند مواد یا مواد فعال، شیمیدان ها باید مناسب ترین روش را برای تولید ترکیب هدف از تعداد زیادی واکنش و روش شیمیایی شناخته شده انتخاب کنند. برای این کار عوامل متعددی مانند بازده محصول مورد نظر و همچنین جنبه های زیست محیطی و ایمنی را در نظر می گیرند. بنابراین توسعه واکنش های شیمیایی جدید و همه کاره همچنان در کانون تحقیقات شیمیایی فعلی قرار دارد.
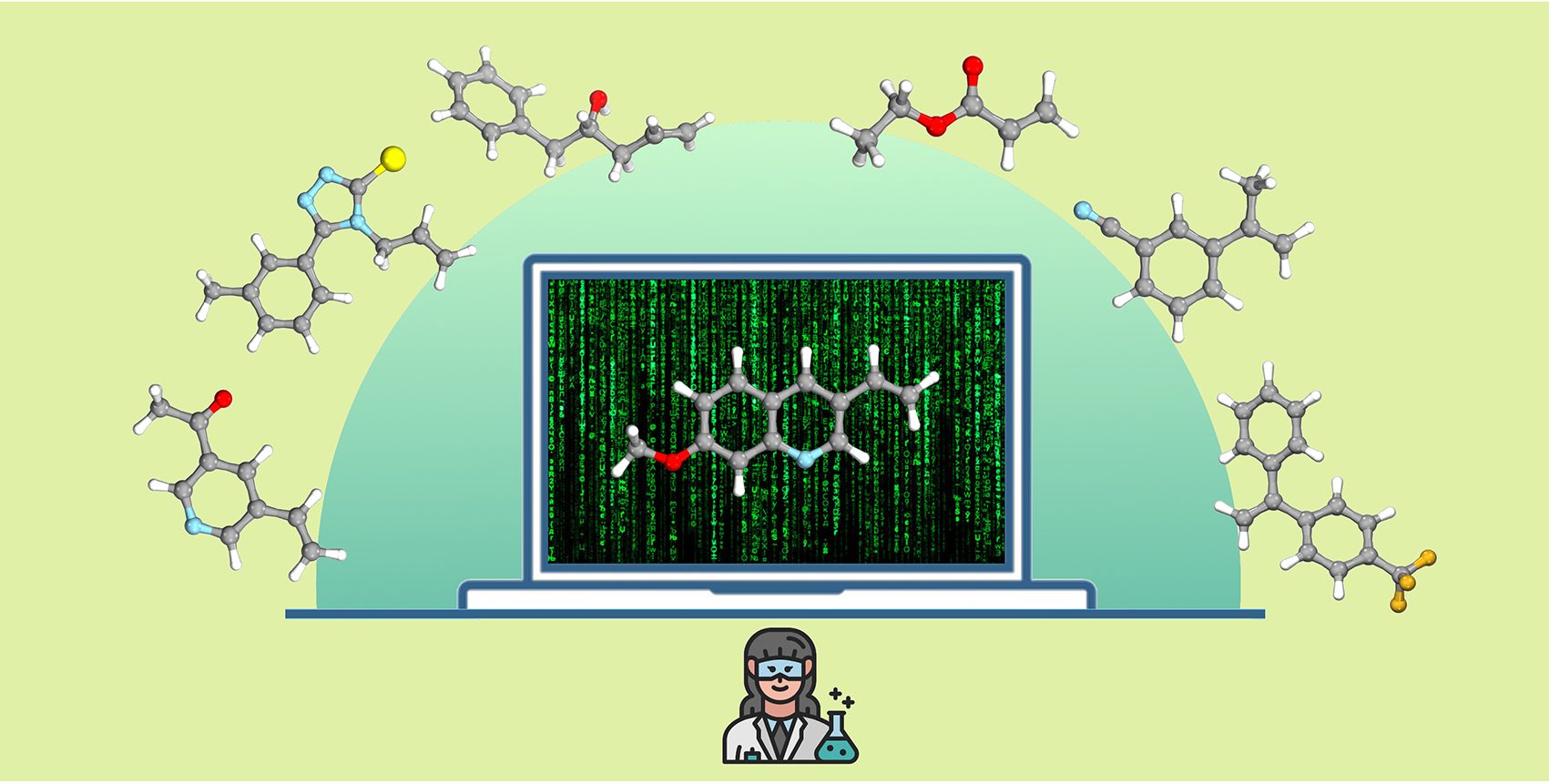
روشی که توسط تیم دانشگاه مونستر توسعه داده شد، از اثر انگشت مولکولی برای انتقال تمام مواد فعال دارویی تایید شده به یک کد دیجیتال استفاده کرد. آنها با استفاده از روشهای یادگیری ماشینی و خوشهبندی بدون نظارت، مدلی را ایجاد کردند که این “فضای” مواد فعال دارویی را به مناطق شیمیایی معنیدار بر اساس ساختارهای مولکولی تقسیم میکند. برای ارزیابی یک واکنش شیمیایی جدید، هزاران بستر آزمایشی بالقوه را می توان با استفاده از مدل یادگیری ماشینی در یک فضا پیش بینی کرد. یک بستر آزمایشی به طور خودکار از مرکز هر یک از مناطق شناسایی شده قبلی انتخاب می شود تا کل فضا را بدون تعصب پوشش دهد.
.Materials provided by University of Münster. Note: Content may be edited for style and length